Matrix Requirements and Galen data join forces to become Matrix One


What is ISO 14971 Risk Management for Medical Devices?

In this blog series, we dive into the specifics of ISO 14971, the pivotal standard governing risk management of medical devices. Whether you're a seasoned professional or a newcomer to the field, our goal is to provide a clear understanding of how ISO 14971 shapes the safety protocols crucial to medical device risk & the development and maintenance of secure medical devices & technologies.
In general, risk can be defined as "the possibility of something bad happening". During our daily lives, we constantly evaluate the risks of different things that come on our path, without even thinking about it.
When we take the car to go to work, we rarely consider that an accident might happen. Even though accidents happen every day. We make an estimation of how likely it is that indeed something bad will happen to us while using our car and consider that the risk of using the car is less than the benefit of using it.
In the medical device field, risks are defined as "the combination of the probability of occurrence of harm and the severity of that harm".
Harm in this case means "physical injury or damage to the health of people or damage to property or the environment".
When developing a medical device, a requirement is to perform a risk analysis. Risk analysis means "a systematic use of available information to identify hazards and to estimate the risk".
Hazard means "potential source of harm"
So bringing all of this together, performing a risk analysis means that you adopt a systematic approach, where based on available information, you identify potential sources of damage to health/property/environment (device risks) and estimate how likely this is to happen and if it happens how bad the result could be.
Looking at these definitions, it's clear that performing a risk analysis is not a one-time shot. It's a continuous process as the available information will change over time for your medical device.
What is Risk Management?
Risk management in ISO 14971 is a systematic process designed to identify, evaluate, and control potential risks associated with medical devices throughout their lifecycle. Risk management is a defined process for identifying potential hazards and controlling or reducing them. A risk management process with your Medical Device should start simultaneously with the design process to minimize potential hazards from the beginning.
The process continues throughout the Medical Device's lifecycle and includes provisions for reviewing post-production data. New information about the product, can lead to the identification of new risks which might have a direct input on the design.
The ultimate goal is to ensure that the benefits of using the Medical Device keep outweighing the risks of using the Medical Device and you reduce the device risk of the medical device.
What are the benefits of ISO 14971?
ISO 14971 is the harmonized standard for medical devices when it comes to risk management. It's current version is the 2019 version and it is applicable to all medical devices.
This international standard outlines the principle and processes for applying risk management to medical devices. It's an internationally accepted framework for manufacturers to integrate risk analysis and risk controls into the entire product lifecycle.
Having one framework helps both manufacturers as well as regulatory bodies to be aligned on how to address and assess risks and risk management in the medical device field, with the ultimate goal of enhancing patient safety and product effectiveness.
The ISO 14971 Risk Management Process
The risk management process consists of
Risk Management Plan
Risk Analysis
Risk Evaluation
Risk Control
Evaluation of overall residual risk acceptability
Risk management report
Analysing and integrating production and post-production information of Medical Device
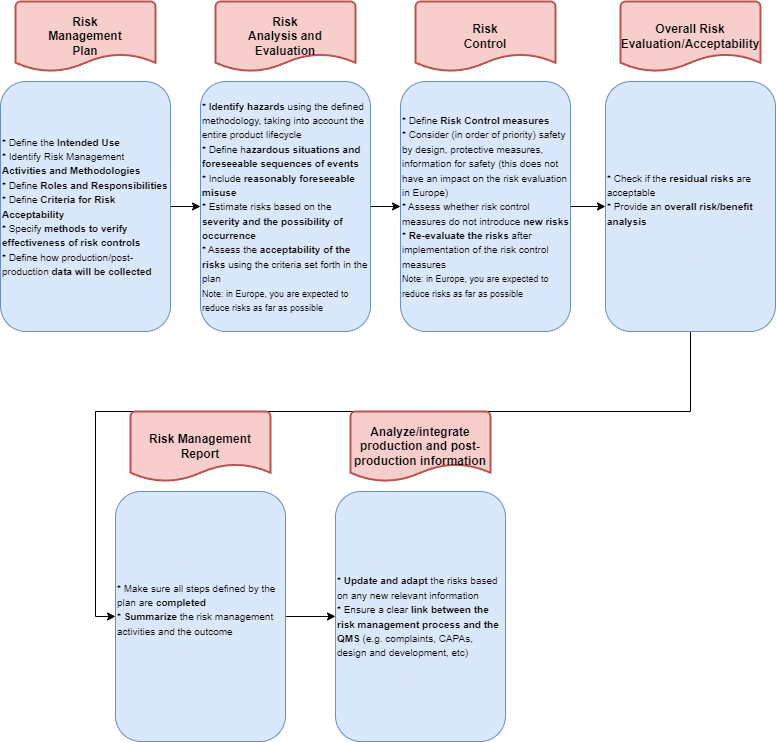 Risk Management Plan
Risk Management Plan
A Risk Management Plan (RMP) in the context of ISO 14971 is a critical document that outlines the approach a medical device manufacturer will take to identify, assess, and control risks associated with their product throughout its lifecycle.
It defines the products to which it applies, the intended use of those products, but as well who will be involved in the risk management activities and what are their respective roles and responsibilities in this process. Furthermore it also describes the methods used to analyse the risks and the different criteria for acceptance.
The foundation for all subsequent activities is laid by the risk management plan.
Risk Analysis and Evaluation
In accordance with ISO 14971, the analysis of risks relies on the device's intended use and safety-related characteristics, which must be clearly defined before initiating the risk analysis process. The initial step involves the identification of potential hazards associated with the device. You can consider hazardous situations linked to these hazards, enabling the estimation of risks associated with each specific hazardous situation.
Sine examples of methods used to identify risks within ISO 14971 are:
Preliminary Hazard Analysis
Fault Tree Analysis
Failure Mode and Effects Analysis
Hazard and Operability Study
Depending on the type of product, the available resources and where you are in the lifecycle of your device, you might adopt one or more of these methods. ISO 14971 does not require you to adopt a specific method for risk identification.
Once you know which hazards, hazardous situations, sequence of events and harms can occur, you can estimate the probability of occurrence and the severity. Together they can give an estimation of the risk. Also here, there are several possibilities. You can do a quantitative or a qualitative evaluation of your risk. You could as well include more parameters such as e.g. detectability. Your risk management plan should define which parameters you will use and how you will make your calculations/estimations.
It could be that over time this will change as you might have more statistical data which allows you to do a more accurate estimation of your risks.
Risk Control
Following the guidelines set by ISO 14971, once risks are defined and assessed for your Medical Device, it becomes essential to minimize them. This involves the implementation of measures outlined in the risk management plan to ensure that the identified risks are effectively mitigated and brought under control.
There are 3 ways to do so:
Safety by design: this is the most profound way of reducing a risk, by implementing changes to the design
Protective measures: in this case, additional protective measures are added to the device or process
Information for safety: here, the user is being informed about certain risks or trained on how to avoid them. This measure has the least impact and should be considered or added in addition if the other two are not possible or insufficient
Note that the aim should be to reduce the risks as far as possible (this is a requirement under the MDR).
When implementing risk control measures, it is important to assess whether these measures do not create new risks. If so, these need to be analyzed and controlled as well to ensure the device risk of the medical device is assessed correctly.
Overall Risk Evaluation/Acceptability
Once all risks have been defined, assessed and risk control measures have been defined and evaluated, an overall risk evaluation needs to be performed for your Medical Device and to reduce device risk. It is the manufacturer's responsibility to make a judgement about the overall remaining risk and to do a risk/benefit analysis in order to conclude whether or not the device's benefits outweigh the remaining risks.
Risk Management Report
Linked to ISO 14971, the Risk Management Report involves the outcomes of the risk management process. It compiles a summary of the risk-related activities, risks, their evaluations, and the corresponding control measures, including their impact for your organization and the medical device. Additionally, the report encompasses an overall risk assessment and draws conclusions regarding risk acceptability and the analysis of risk/benefit.
Risk Management File
The Risks Management File is the part of the technical documentation of the medical device that contains both the Risk Management Plan as well as the Report and possible attachments to the risk management process.
Production and Post-Market Information
In alignment with ISO 14971, the Risk Management Process is an ongoing effort. As the device transitions from development to manufacturing and enters the market, new insights become available. Manufacturers are required to actively and passively engage in post-market surveillance, conduct clinical evaluations, and systematically collect information for thorough data analysis. Feedback obtained through this process should seamlessly integrate back into the design and the risk management process. Given that risks may evolve over time, continuous vigilance is important. Regular updates to risk assessment and evaluation are essential whenever there are changes in the available information. This iterative approach aligns with the principles outlined in ISO 14971, ensuring the ongoing efficacy of the Risk Management Process.
Support Risk Management with QMS
The Risk Management process is not something that stands alone and is highly connected to other processes within the Quality Management System, such as design control, complaints handling, CAPAs, etc.
A system that allows to link your CAPAs and complaints directly with your risks and your design without having to duplicate work, can be a real timesaver.
MatrixQMS and MatrixALM provide the possibility of having your QMS and design documentation in one platform. The system allows you to analyze the risks as you have defined in your risk management plan.
Being able to define your risks and link them immediately to your design, gives a clear overview of what risks are controlled properly and where are possible gaps. Our focus on traceability in design allows you to make sure all of your risk control measures are properly implemented and tested.
If you are interested in learning more how we can help you in performing risk management in an agile way, reduce device risk of your medical device, aligned with ISO 14971 requirements, don't hesitate to reach out to us for a demo!
ISO 14971 Video guide
Learn more with our latest video guide for ISO 14971 and how you can prepare your Risk Management processes for your medical Device.