Matrix Requirements and Galen data join forces to become Matrix One


Agile Compliance - The 9 most important terms of IEC 62304 and what they mean for Agile Developers

Achieving compliance with IEC 62304 while embracing the agile software development process can be a challenge for any developer. The agile methodology often clashes with the requirements outlined by IEC 62304 for developing medical device software.
In this article, we will explore the 9 biggest most important terms of IEC 62304 and the impact on agile development. By understanding these gaps, Medical Device suppliers can bridge the gap and navigate the complexities of regulatory compliance while benefiting from the benefits of agility.
Plans, Records & Reports
These are actually the most important artifacts to be produced to demonstrate compliance.
Some typical documents an auditor may request during an audit:
Software Development Plan
Software Verification Plans
Software Usability Engineering Plan
Risk Management Plan
Product Validation Plan
Product User Requirements Specification
Software Requirements Specification
Verification Report
Validation Report
Risk Management Report
Records: Minutes of meetings, Project Kickoff, Release Note, and Final Design Review, Reviews for different milestones of the development process
These documents need to be controlled, e.g. dated, signed, stored and versioned in dedicated locations. They will be the central part of your Technical File (for European submissions) and/or Design History File (for the US).
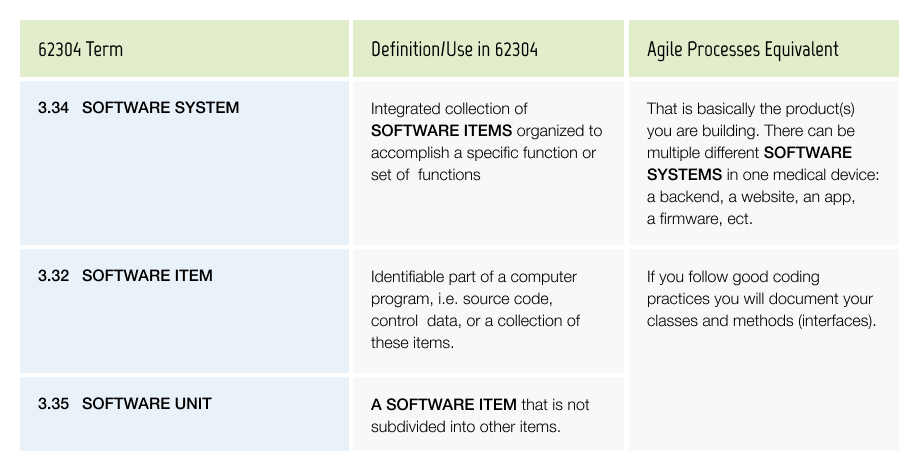
 Software System, Item & Unit
Software System, Item & Unit
All software systems of your device and their relationships to each other need to be included in your Technical Documentation (Design History File or Technical File). The idea of the standard is that you document how the SOFTWARE SYSTEMS you build are decomposed into a tree of SOFTWARE ITEMS with some SOFTWARE UNITS as leaves. The level of detail is up to you, but it needs to be clear to the auditor that it is detailed enough for a risk analysis and planning of testing activities.
Keep in mind: This means auditors are not interested in auto generated class documentation of your product. Specifically as developers are supposed to do the architectural design before the implementation.
 Software of Unknown Provenance (SOUP)
Software of Unknown Provenance (SOUP)
Implementing particular processes (See Season II Episode 5) & recording specific information about SOUP/OTS or legacy software, is important if you wish to remain compliant.
Some key examples of this information are: Manufacturer details, Version of the released product, Known issues, Risks.
 Software Safety Classification & Segregation
Software Safety Classification & Segregation
As the Software Safety Classification can have a large impact on the amount of documentation and required work, it needs to be determined and documented as early as possible (See Season II Episode 2).
Segregation allows teams to have different software safety classes in different Software Systems. If the architecture can be done in such a way that only a small part of the software has a high risk class, it can save a tremendous amount of documentation work.
 Traceability
Traceability
For a medical device, documenting Traceability between design input (what a user needs) through several levels of design output: what does the product do, what does the software do, how has it been tested, and what are the associated risks is a required practice.
The Traceability needs to be complete and accurate, showing exactly what the design input of the released product is and how it links to the design output up to the verification & validation.
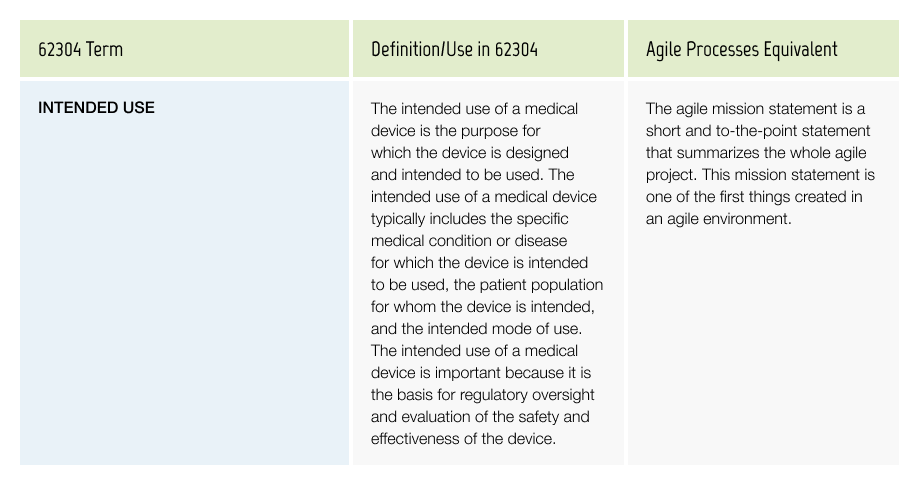
 Intended Use
Intended Use
The intended use of a medical device is a description of the specific medical purpose for which a device is intended to be used.
 Useability
Useability
There is a specific standard (IEC 62366) which requires you to document how you ensure the usability of the product (See season III).
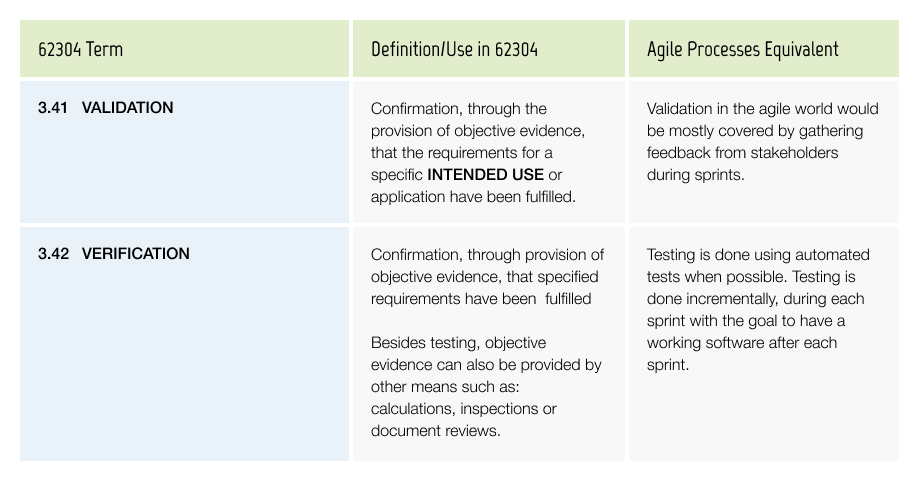
 Validation & Verification
Validation & Verification
Validation is the proof that the product does what it was created for. While it is usually done on a product level, it also covers the software of the medical device. If your device consists of only software (SaMD) you are obliged to validate it as described in IEC 8203. From a regulatory standpoint, verification is the proof that the software works reliably against what was specified.
Traceability is required from the design input (e.g. user, software or regulatory requirements) to the executed validation and verification tests. Proof that the tests cover the whole product and all its functions and that they pass for the released software version(s). The tests need to run on the version of the software which is released and the documented test results need to contain all relevant details, e.g. who executed the test on which version, what were the results (evidence that the step has been executed successfully) for each test.
 Quality Management
Quality Management
The quality system of the company has an impact on many aspects of the software development process.
Examples include: How to create, store and sign documents.
Risk management is the central component of medical device regulations. This has a large impact during the software development and deployment phases.
 Key Takeaways
Key Takeaways
Developing software for medical devices can be done using agile processes but it's important to make sure that the documentation created during development conforms to the standards. Therefore, it is important that Developers understand the terms explained above, so that you can tailor the agile process to meet the regulatory needs.
You need to create Plans, Records & Reports of your software development activities, inputs and outputs, complete the traceability from the kickoff to verification & validation of the Medical Device. Documenting your software architecture as software system/items and units, whilst optimizing the software safety classes assigned to each using segregation is no easy feat, however it’s necessary if you want your Medical Device to pass an audit.
IEC 62034 is not the only standard with an impact on agile software development, there are other standards for Risk Management, Quality Management, Cyber Security, Useability and more. Stay tuned for our next season where we’ll dive into these topics, and more!