Matrix Requirements and Galen data join forces to become Matrix One


Why does managing software requirements for medical devices differ from other industries?

How does requirement management for Medical Devices is different to other industries?
When designing the software for medical devices, the top priority is the safety of the patient, physicians and other involved people. In other, less regulated, industries that might be very irrelevant.
But this is not all which is different, because when designing a medical device you need to keep in mind the time a user (patient or physician) actually has to use the device. How long does a doctor have for an intervention with a patient? A few minutes max and most of the time (s)he will actually talk to the patient, so what is left for the software? Almost nothing...
So the goal of the design is to make the most simple user interface possible, not the most powerful software: the user is most likely to only ever use very few functions. I have seen ultra sound machines where the user could change image parameters in several layers of settings. But a doctor might only ever use 2 different combination of settings.
How to come up with the right amount of requirements?
If you work with user stories, define your personas well: who will actually use the device?
Once you decided what your users and their use cases are, ask yourself: What are the two things they actually want to achieve within the 5 seconds they have to use your device? For every other feature or story, ask yourself when would a user really have the time to use it.
Once you know the essential things a user needs to do, think how it can be done most efficiently and safely.
For example for ultrasound machines, there are usually two use cases: either for emergency or scheduled. If they are used in an emergency situation they could be used to scan whatever part of the body, for the doctor it's less important to have his preferred image settings as long as the image is good enough for a first assessment. In the routine use however, the doctors will only scan a few organs and once they have their preferred settings they won't try to optimize them while they examine a patient.
How to document the requirements within the context of IEC 62304?
Since you develop a medical device there are a few things you need to do more formal than for other software projects:
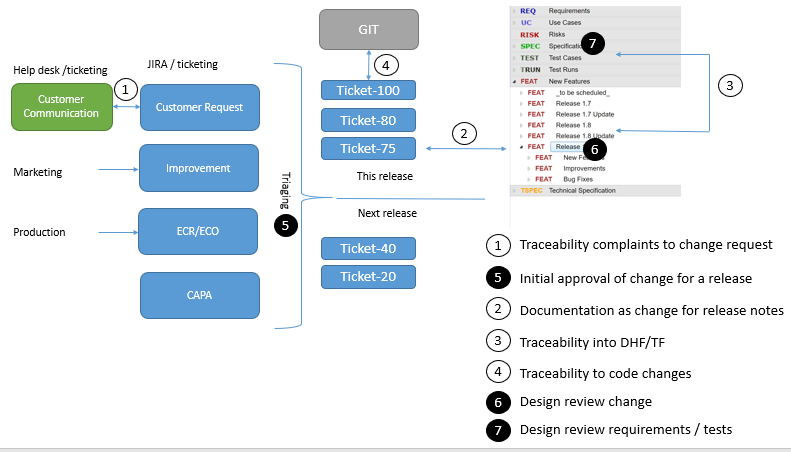
You need to know where your requirements come from (are they risk controls, based on customer complaints, internal ideas for improvement)? This can be achieved using traceability between your requirement and the complaints or the risk for example.
You need to know in which release or product a change has been done. Again this can be achieved using traceability.
You need to communicate changes to users. So you need per release to be able to easily create release notes with all relevant changes
You need to review and approve changes before they are done. This also needs to be documented.
You need to know which change causes which changes in the source code. Again this can be managed by traceability.
Cybersecurity is more and more important key during the design, even if the software of your device does not contribute a direct risk to a patient, the patient data needs to be protected by all means.
How can this be achieved most easily?
We propose to keep a backlog of possible changes in an issue tracking system (such as e.g. JIRA).
In your SCM (software configuration management tool, like git or svn) make a rule that every commit required a change ID (e.g. the jira ticket).
Complaints should also go in a database (e.g. JIRA service desk) and be processed according to your procedures (e.g. triggering CAPAs). If they require a software change they should be linked to another item in the backlog.
Changes in the backlog can be prioritized and (according to your procedures) assigned to developers, documenting the decision that something should be implemented. At the same time, a change item should be created in a category document all your changes in Matrix Requirements (unless it is a bug fix for another change which is not yet released). This change item should be formulated to be usable for the release notes and it should be formally reviewed (e.g. to ensure that all related risks have been documented) and approved. In Matrix Requirements, there are several options you have to perform these reviews.
Once the change has been reviewed, it should be linked to your design input or output. Often a new feature will be a new top level requirement, an improvement might just be a change of a specification and a bug fix might link to an improved test case.
If you want more details on how to control changes to SW in medical device software and how our tool, Matrix Requirements, can help you with that, don't hesitate to contact us.